中国罕见病患者的春天来了:国家关注、社会支持、制度出台
2008年2月29日,欧洲罕见病组织发起了第一届“国际罕见病日”。选择这个日子,是因为这是每四年才会出现一次的日子,寓意:“罕见”。之后,每年的2月最后一天被定为“国际罕见病日”。今年,罕见病日的主题是“搭建健康与关爱的桥梁”。

他们所患的疾病,病名听起来都很陌生,离我们仿佛也很远;他们的遭遇,越来越受到人们的关注;他们就是罕见病患者。如今,罕见病及罕见病患者越来越受到人们的关注。长期致力于罕见病研究的北京大学第一医院丁洁教授说,罕见病多学科的诊治和研究在发达国家已经有几十年的历史,许多重大的研究成果都是从罕见病的研究开始。最近几年,随着精准医疗和个体化治疗的医学模式发展,众多世界著名的医院、医疗研究机构和制药企业纷纷进军罕见病领域。10年前,“罕见病”一词在我国是真的“罕见”,无人知晓,少人问津。然而,经过多年的努力,罕见病终于站到了聚光灯下,国家关注、社会支持、制度出台。尽管罕见病诊疗领域依然面临着各种挑战,但罕见病患者们的春天来了。
脊髓型肌萎缩症患儿草根:
和同龄孩子一样读小学四年级
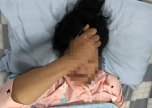
9岁男孩草根,是一个脊髓性肌萎缩症患儿。9年前,草根刚刚6个月时,妈妈发现他不会踢腿。细心的妈妈带着草根走上了就医之路。经过几个月的排查,草根最终被确诊为脊髓型肌萎缩症。病虽然确诊了,但妈妈得到了这样的反馈:“病没法治,也没药。”
脊髓性肌萎缩症是一种罕见的遗传性神经肌肉疾病,新生儿发病率大约为万分之一,也是导致婴儿死亡的重要遗传因素。脊髓性肌萎缩症的典型症状就像病名一样:肌肉萎缩。因为肌肉萎缩,患者的运动功能会严重受限,甚至连普通的翻身、蹬腿、爬行都难以实现。随着病程的发展,肌肉逐渐萎缩,最终影响吞咽和呼吸,严重威胁患者生命。
草根的妈妈说,当时,脊髓性肌萎缩症的相关文献以及资料特别少,医生也没有办法。但她觉得事情没有这么绝对,妈妈找了康复师,按照正常孩子的教育方式来教育儿子。不轻易放弃的草根妈妈,终于见到了自己努力的成果:9岁的草根现在和同龄孩子一样读小学四年级,不仅学习成绩优异,而且在英语比赛多次获奖。草根喜欢下国际象棋,还获得了“国家四级棋士”。草根活泼开朗,笑容和小时候一样灿烂。“我想成为国际象棋的国家大师,如果可以走路,就去当兵保卫祖国!”
依据发病年龄和以及患儿保留的运动功能程度,脊髓性肌萎缩症从重到轻被分为0、I、II、III、IV五个类型。0型患者病情最重,患儿出生后就会发病,不能自主呼吸。如果不进行治疗,大多数患有I型患儿甚至活不到两岁。草根属于II型患儿,这些孩子通常在出生后6到18个月内发病,孩子能够坐起来,可以写字,如果护理的好,可以活到成年。
脊髓性肌萎缩症患者是罕见病,但在人群中的致病基因携带率非常高,大约2%左右,也就是50个普通人中就有一个是致病基因携带者。一旦夫妻双方同为致病基因携带者,生下脊髓性肌萎缩症的孩子概率就达到25%。这些患儿非常不幸,但欣慰的是,由于致病原因较为明确,目前脊髓性肌萎缩症在诊断、护理和治疗上都有了很大发展,也有了治疗药物。去年9月,全球唯一的治疗药物作为已经在境外上市且临床急需的罕见病治疗新药,被国家药品监督管理局纳入优先审评审批程序。草根们看到了希望。
戈谢病患者小雷:
成为互联网行业工作人员
29岁的小雷是一名互联网行业工作人员。平时他上班下班,和同龄人一样。但是,他的童年是很多人难以想象的:小雷是一名戈谢病患者,他的弟弟也是一名戈谢病患者。
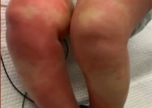
“弟弟5岁时,终于确诊了。我的症状虽然比弟弟轻,但症状完全一样,不用检查就直接确诊了。”戈谢病是一种罕见遗传性的代谢疾病,患者由于体内缺乏葡糖脑苷脂酶,表现为生长发育落后,肝脾肿大,贫血,患者往往多器官功能损伤,甚至会危及生命。三分之二的戈谢病患者在儿童期发病。小雷和弟弟当时也出现了肝脾肿大的情况,“肚子大得就像个皮球。”弟弟当时肝脾肿大格外严重,由于当时还没有药物可以治疗,医生建议先治标,“切除脾脏”。弟弟上了手术台,结果因为麻药过敏,又从手术台上下来了。
兄弟俩的症状起起伏伏,不仅有脾脏肿大、骨痛等情况,还有一个小麻烦:流鼻血。“别人流鼻血几分钟就止住了,我们流鼻血,不知道要什么时候才能止住。”上小学时,学校就在家附近,流鼻血后小雷就会跑回家,家人来照顾他。上初中时,由于学校离家太远,流鼻血之后没有家人照顾,小雷不得不两次中止了学业。幸运的是,就在这一年,已经在国外上市的酶替代治疗药物通过世界健康基金会的援助来到国内,为包括小雷和弟弟在内的130名戈谢病患者提供无偿药品援助,这一援助就是20年。治疗药物是一种注射剂,第一针打下去之后,小雷再也没有出现过长时间流鼻血的情况;一年之后,肿大的肝脾开始回缩;三到四年后,困扰小雷的骨痛症状也消失了,小雷和弟弟的生长发育也没有受到影响。
治疗戈谢病的药物,每一针的价格是2.3万元,一个月要注射7支,一年的费用就是200万。如果没有援助项目,小雷的父母完全不能承受两个孩子的费用。几经变更,当时的援助项目的合作方现在变更为中华慈善总会。2009年,该项目正式更名为思而赞慈善援助项目。在援助项目的支持下,小雷和弟弟都健康地读完大学,如今都正常工作。不同的是,他们每周要给自己注射一针,每4个月要领一次赠药。“我们曾经在深渊里苦苦挣扎,是科技进步和援助项目,带着我们两个跳了出来。”
国家出台相关政策
保障2000多万罕见病患者用药
近年来,国家针对罕见病接连出台了多项医疗惠民政策。罕见病领域的各学科专家们团结起来,成立了罕见病学术组织、多学科合作诊治模式,逐渐建立了罕见病的诊疗体系,发表相关专家共识、“研究报告”,积极地宣讲培训人才,更坚持开展科研工作去更深入地理解和治疗罕见病。但是,在丁洁教授看来,“前方依然任重道远。罕见病种类繁多,临床表现复杂多样,导致诊断难、治疗难,国内医生对罕见病的诊治尚缺乏足够的认识,常常导致患者难以得到及时诊治。”从治疗领域来说,罕见病的规范诊疗问题,也是影响罕见病诊治水平的重要原因。如何让罕见病患者得到及时、正确的诊治,如何为政府的政策制定提供可靠的相关数据;如何推进罕见病临床和基础研究,这些任务都需要进一步努力完成。当然,还有更多患者需要接受规范治疗,其中还涉及罕见病药物制度和医疗保障体系的完善等相关问题有待于不断探索解决。
现在,越来越多的罕见病患者及家庭看到更多的希望。2018年,国家五部委联合发布《第一批罕见病目录》,共有121种疾病被收录其中,这被业内视为罕见病保障历史性的突破。今年2月11日,国务院常务会议中明确指出,要保障2000多万罕见病患者用药。自2019年3月1日起,国家对首批21个罕见病药品进口环节减按3%征收增值税,国内环节可选择按3%简易办法计征增值税。2月15日,国家卫健委发布《国家卫生健康委办公厅关于建立全国罕见病诊疗协作网的通知》,遴选出了罕见病诊疗能力较强、诊疗病例较多的324家医院作为协作网医院,组建罕见病诊疗协作网。丁洁说,尽管罕见病的治疗等方面还面临着诸多挑战,但无论是诊疗手段、社会认知及关注、还是国家政策等方面,罕见病领域已发生令人瞩目的变化,“这些改变还将继续,罕见病患者已经迎来了春天。”
来源 北京晚报 记者 贾晓宏 编辑 丁肇文
流程编辑 TF003