12岁女孩患罕见病成了“粘宝宝”,看着像五六岁只吃软食流食
在中国,由于庞大的人口基数,万分之一、十万分之一发病率的“罕见病”,也变得并不罕见了。2月28日,是国际罕见病日。

重庆女孩哆哆(化名)今年12岁,身高只有1.17米,看上去就像五六岁的孩子。“在他们那群娃儿里头,她算是长得高的,很多只有80、90厘米。”郭爸爸说。8年前,郭爸爸从医生那里听到一个新词——“粘多糖贮积症”,中年得女的郭爸爸和哆哆都被这个词改变了命运。“都说这个病活不过10岁,现在(哆哆)熬过了,努力再熬20岁。”
哆哆4岁的时候,郭爸爸发现女儿的手肘有些不一样,像是脱臼了、骨头突出来,在重庆的医院,医生第一次质疑性地提到了“粘多糖贮积症”,随后,郭爸爸带着女儿到北京求医,经过基因检测,最终确诊。
这意味着,哆哆是个“粘宝宝”。这是对粘多糖贮积症孩子的爱称,因为他们大多身材矮小,所以被冠以“宝宝”的称谓;同样也因为“长不大,活不长”是很多医生对黏多糖孩子的残酷论断,所以被称为长不大的粘宝宝。一般情况下,患儿多在10岁左右死亡。
哆哆确诊第一个月,郭爸爸睡不着觉,一夜之间白了头。
四川大学华西第二医院小儿遗传代谢内分泌科副主任吴瑾介绍说,粘多糖贮积症是一种基因遗传病。粘多糖贮积症是因为遗传了父母相同的缺陷基因,造成体内一种酶缺乏,直接导致不能分解体内的黏多糖(硫酸皮肤素和硫酸乙酰肝素),从而堆积在体细胞内并造成全身器官系统的损伤。简单地说,粘宝宝们的身体就像一台发动机,由于机油中有不断有渣滓沉积无法分解,所以发动机开始运作不畅、机身受损,直到完全罢工。
8年求医,郭爸爸的手机里存着各种粘多糖贮积症的资料。他知道了女儿的粘多糖症属于四型,还知道粘多糖症一共有七种分型、很多种亚型,每个粘宝宝的病症都不一样,“毒素”侵蚀到哪里,哪些器官就开始出问题。有的孩子会有智力障碍、心血管疾病,有的听力、视力受损,“我家的智力很正常,一直都在读书,成绩还挺好。”
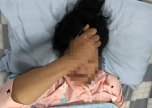
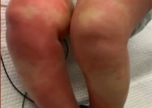
但4岁起,哆哆的手臂耻骨就没有发育,桡骨正常生长形成畸形,X型腿,颈椎骨骼没有发育完全,有瘫痪风险。前几年,哆哆还能去操场,还能走出几百米,如今,只能蜷缩在小轮椅中。已经12岁的哆哆,只能吃下软食和流食,稍微硬一点,就会哽住,“像照顾一两岁的娃儿。”3月2日,华西第二医院举行罕见病义诊,郭爸爸带着哆哆来请骨科医生看看,“钙流失得很严重。”
吴瑾介绍说,粘多糖贮积症并没有很好的方法可以完全根治,在国外有获批的酶替代疗法,可帮助减轻患者的病情、舒缓痛苦和不适,以延长他们的生命。但即使有药,一年高达数十万至百万的治疗费,对于患儿家庭来说,也是无比沉重的负担。这是罕见病难确诊、易漏诊之外的,最大的影响因素。
来源:红星新闻